What is cementoma
This is a type of benign tumor that forms in the area of the tooth root. It consists of fibrous cement-like tissue and at first does not manifest itself in any way - only a specialist can diagnose the problem. The development of the pathological process is accompanied by the gradual destruction of bone and epithelial tissue in the oral cavity. When the tumor reaches a large size, deformation of the jaw occurs. In some cases, the disease can lead to a fracture of the jaw bone. Women, as well as persons under the age of 20, are more susceptible to pathology.
Only a specialist can diagnose the problem
Most often, cementoma forms in the area of the central teeth of the lower jaw. In other cases, the tumor may be localized in the upper jaw, but this situation is especially dangerous, since here the pathological process can affect the maxillary sinuses. According to ICD-10, cementoma belongs to category D16 - benign neoplasm of bones and articular cartilage, subtype D16.4 - bones of the skull and face.
Publications in the media
A huge number (hundreds!) of nosological units with the generic word “Dysplasia” are known. This article lists in alphabetical order those nosological units that could not be placed in other articles in the reference book characterizing dysplasia (Craniofacial dysplasia, Ectodermal dysplasia, Epiphyseal dysplasia, Dental development disorders, Chondrodysplasia, Achondrogenesis). Many dysplasias, like the vast majority of genetic diseases and phenotypes, are also difficult to identify using the ICD-10 system.
Acromicric dysplasia (102370, В), congenital acromicria. Clinically: moderate facial anomalies, shortening of the hands and feet, severe growth retardation, short metacarpal and phalangeal bones. Laboratory findings: disorganized cartilage growth. ICD-10. Q87.1 Syndromes of congenital anomalies manifesting predominantly as dwarfism
Arterial fibromuscular dysplasia, see Fibromuscular dysplasia.
Diastrophic dysplasia - skeletal dysplasia with severe curvature of bones • Diastrophic dysplasia (222600, 5q31–5q34 5q32–5q33.1, mutations of the transmembrane sulfate transporter gene DTD, r). Clinically: congenital dwarfism with short limbs, impaired ossification and congenital epiphyseal cysts, hypertrophy of ear cartilage, cleft hard palate, kyphosis, scoliosis, abducted thumb, fusion of proximal interphalangeal joints, brachydactyly, bilateral clubfoot, calcification of rib cartilage • Pseudodiastrophic dysplasia ical (264180 ). Clinically: rhizomelic shortening of the limbs, interphalangeal and metacarpophalangeal dislocations, elbow dislocations, severe clubfoot, increased distance between the coronal sutures of the skull, hypoplasia of the middle third of the face, hyperthermia, platyspondyly, tongue-like deformities of the lumbar vertebrae, scoliosis, hypoplasia of the 2nd vertebra, pronounced lumbar lordosis • Congenital bone dysplasia de la Chapelle (#256050, r). Clinically: Lethal at birth, severe micromelia, kyphosis of the cervical spine, clubfoot equinovarus, abducted hallux, abducted toes, duplication of the middle phalanges, cleft palate, patent foramen ovale, respiratory failure, laryngeal stenosis, softening of the cartilages of the larynx and trachea, hypoplasia lungs, shortness of breath, small chest, congenital bone dysplasia, triangular fibula and ulna, platyspondyly, pathological metaphyses and epiphyses, sacral anomalies, additional pelvic ossification points. Laboratory: lacunar halos around chondrocytes in skeletal cartilage. ICD-10. Q77.5 Diastrophic dysplasia.
Oculomaxilloosseous dysplasia (*164900, Â). Corneal opacity and multiple anomalies of the lower jaw and limbs. Synonym: OMM syndrome (from: o phthalmo m andibulo m elic). ICD-10. Q78.8 Other specified osteochondrodysplasias.
Greenberg dysplasia (215140, r) is congenital lethal dwarfism. Clinical picture: dwarfism with short limbs, prenatal death, severe fetal hydrops, noticeably shortened, “moth-eaten” long tubular bones, unusual ectopic ossification points, pronounced platyspondyly, pronounced extramedullary hematopoiesis. Synonym: hydropic chondrodystrophy. ICD-10. Q77.1 Small stature incompatible with life.
de Morsier dysplasia (septo-optic dysplasia, 182230, Â?). Hypoplastic optic discs with a double edge, absence of the septum pellucidum, GH deficiency, pathology of the corpus callosum and cerebellum. ICD-10. Q04.4 Septooptic dysplasia.
Diaphyseal dysplasia (Engelmann's disease) is a progressive symmetrical hyperostosis of the diaphysis of long tubular bones from the periosteum and endosteum with sclerosis of the newly formed bone tissue. Clinically: asthenic physique, severe pain in the bones of the legs, fusiform swelling of the lower leg, multiple subungual hemorrhages, myopathy, waddling gait, compression of cranial nerves, weakness, muscle fatigue, scoliosis, lumbar hyperlordosis, hypogonadism, anemia, leukopenia, increased ESR, hepatosplenomegaly, onset aged 10 to 30 years, sensitivity to GC, dysplasia, osteosclerosis and hyperostosis of the diaphysis. Synonyms •• Camurati-Engelmann disease •• Ribbing's disease •• generalized hyperostosis •• systemic diaphyseal congenital hyperostosis •• progressive diaphyseal dysplasia •• systemic hereditary osteosclerosis with myopathy. ICD-10. Q78.3 Progressive diaphyseal dysplasia.
Dissegmental dysplasia is a group of hereditary skeletal dysplasias manifested by dwarfism, damage to the brain and internal organs. At least 2 forms, differing in clinical, radiological and morphological signs • Handmaker-Silvermann dissegmental dysplasia (224410, r) - lethal form. Clinically: vertebral bodies of various sizes and shapes, early death, the clinical picture resembles Kniest syndrome • Dissegmental Rolland–Debuquois dysplasia (224400, r) is a milder form. Clinically: congenital chondrodystrophy, dwarfism, abnormal segmentation of the vertebrae, limited joint mobility, micromelia, curvature of the limbs, high palate, cleft hard palate, hydrocephalus, hydronephrosis, hypertrichosis. Synonyms: dissegmental dwarfism •• anisospondylic campomicromelic dwarfism •• Rolland-Debuquois syndrome • Dissegmental dysplasia with glaucoma (601561) - the phenotype resembles Kniest dysplasia (156550), and dissegmental dysplasia (224400, 224410), combined with severe glaucoma oh. ICD-10 • Q77.1 Small stature, incompatible with life • Q77.3 Point chondrodysplasia • Q77.5 Diastrophic dysplasia.
Campomelic dysplasia (114290, Â, more often *211970, 17q24.3–q25.1, SOX9 gene [CMD1, SRA1, SRY], r) - congenital lethal dwarfism with short limbs, small cartilaginous skull size, platybasia, hypertelorism, depressed nasal bridge , micrognathia, cleft palate, recessed tongue, pulmonary hypoplasia, tracheal hypoplasia, narrow pelvis, hip abnormalities, platyspondyly, kyphoscoliosis, hypotonia, absence of olfactory nerves, small hypoplastic scapulae, 11 pairs of ribs, short phalanges of the hands and feet, moderate curvature of the femoral and tibial bones, leg equinovarus deformity • Grant family syndrome (138930, Â) is one of the forms of skeletal dysplasias of the campomelic type. Clinically: blue sclera, hypoplasia of the jaws, campomelia, curvature of the clavicles, femurs and tibias, sloping shoulders, additional bones in the sutures of the skull. ICD-10. • Q77.1 Small stature incompatible with life.
Bone dysplasia with medullary fibrosarcoma (112250, BDMF gene, 9p22–p21, r). Clinically: skeletal dysplasia, malignant fibrous histiocytoma, bone fractures with minimal trauma, multiple necrosis of the bone diaphysis, compaction of the cortical layer of the diaphysis. ICD-10. C41 Malignant neoplasm of bones and articular cartilage of other and unspecified locations; C41.8 Damage to bones and articular cartilage, extending beyond one or more of the above locations.
Craniocarpotarsal dysplasia (*193700, Freeman–Sheldon syndrome, Â, r). Clinically: hypoplasia of the nose, mouth, deep-set eyes, ocular hypertelorism, camptodactyly; scoliosis. ICD-10. Q78.8 Other specified osteochondrodysplasias.
Cranio-metaphyseal dysplasia - dysplasia of the metaphyses of long bones in combination with severe sclerosis and thickening of the skull bones (leontiasis ossea), hypertelorism. ICD-10. Q78.8 Other specified osteochondrodysplasias.
Mesomelic Nivergelt dysplasia (*163400, Nivergelt syndrome). Clinically: short limb, dwarfism recognized at birth, radioulnar synostosis, rhomboid tibia and fibula, synostosis of the tarsal and metatarsal bones. ICD-10. Q77.8 Other osteochondrodysplasia with growth defects of long bones and spinal column.
Mesomelic Reinhardt-Pfeiffer dysplasia (191400, Â). Congenital dwarfism, hypoplasia of the bones of the forearm and lower leg. ICD-10. Q78.8 Other specified osteochondrodysplasias.
Metatropic dysplasia (dysplasia) - congenital dwarfism with damage to the metaphyseal cartilages • Non-lethal form (156530, Â) • Lethal form (*250600, r): death in utero or shortly after birth. Clinically: intrauterine growth retardation, relatively short spine, severe scoliosis, kyphosis, anisospondyly, pelvic anomalies, hyperplasia of the femoral epicondyles, abnormal shape of the metaphyses, respiratory failure. Laboratory examination: violation of the formation of cartilage of the trachea and bronchi, absence of spongy substance of the metaphyses. ICD-10. Q78.5 Metaphyseal dysplasia.
Metatropic Knistic dysplasia is a group of hereditary skeletal diseases manifested by rhizomelic dwarfism, probably due to collagen defects (#156550, collagen gene COL2A1 [120140], Â): metatropic dwarfism, macrocephaly, flat face, myopia, retinal detachment, cataract, hearing loss, cleft palate, platyspondyly, inability to clench the hand into a fist. Laboratory examination: pathological collagen of cartilage under electron microscopy, excretion of keratan sulfate in the urine. ICD-10. Q78.5. Metaphyseal dysplasia. OMIM. Metatropic dysplasia: •• type I (*250600) •• type 2 Cnista (#156550) •• with protruding lips and ectopic lens (245160) •• lethal (245190).
Metaphyseal dysplasia. Impaired transformation of the metaphyses of long bones into a normal tubular structure; at the same time, the ends of the long tubular bones become thickened and porous, the cortical layer becomes thinner. ICD-10. Q78.5 Metaphyseal dysplasia.
Metaphyseal multiple dysplasia is a congenital disease characterized by thickening of long tubular bones, valgus deformation of the knee joints, flexion ankylosis of the elbow joints, enlargement and deformation of the skull cranial metaphyseal dysplasia. ICD-10. Q78.5 Metaphyseal dysplasia.
Mondini dysplasia is a congenital anomaly of the bones and membranous ear labyrinth, characterized by aplasia of the cochlea of the inner ear and deformation of the vestibule and semicircular canals with partial or complete loss of auditory and vestibular functions. ICD-10. Q16.5 Congenital anomaly of the inner ear.
Oculoauriculovertebral dysplasia (*257700) is a syndrome characterized by epibulbar dermoid, anomaly of the auricle, micrognathia, vertebral and other anomalies “Goldenhar syndrome. Q18.8 Other specified malformations of the face and neck.
Oculovertebral dysplasia - microphthalmia, coloboma or anophthalmia with a small orbit, unilateral dysplasia of the upper jaw, macrostomia with underdeveloped teeth and malocclusion, malformations of the spine, cleft and underdeveloped ribs. ICD-10. Q87.8 Other specified congenital anomaly syndromes not elsewhere classified.
Otodental dysplasia (*166750, Â) - sensorineural hearing loss, dental anomalies (ball-shaped teeth, absence of small molars, molars with two pulp chambers, taurodontia, pulp stones). ICD-10. Q87.8 Other specified congenital anomaly syndromes not elsewhere classified.
Spondylometaphyseal dysplasia is a heterogeneous group of skeletal diseases with impaired growth and formation of the spine and long tubular bones; it differs from spondyloepimetaphyseal and spondyloepiphyseal dysplasias by involving only the metaphyses of the tubular bones. All three groups of dysplasia have spinal abnormalities. Spondylometaphyseal dysplasias are often observed as isolated cases, but various inherited forms with dominant, X-linked and recessive modes of inheritance have been described. ICD-10. Q77.8 Other osteochondrodysplasia with growth defects of long bones and spinal column. OMIM: Spondylometaphyseal dysplasia: • Goldblatt (184260) •• with angular fractures (184255) •• Algerian type (184253) •• with enchondromatosis (271550) •• Richmond type (313420).
Spondyloepimetaphyseal dysplasia (SEMD) is a heterogeneous group of skeletal diseases with impaired growth and formation of the spine and long bones. SEMD differs from spondylometaphyseal dysplasia (SMD) and spondyloepiphyseal dysplasia (SED) by involving both the metaphyses and the epiphyses. In all three groups of dysplasias (SEMD, EDS and SMD), there are spinal anomalies. SEMD is often observed as isolated cases, but various inherited forms with dominant, X-linked and recessive modes of inheritance have been described • Kozlovsky spondyloepimetaphyseal dysplasia (*184252, Â): short stature, usually manifests between 1 and 4 years of age, short trunk, pathological femoral necks and their trochanter, general platyspondyly • Spondyloepimetaphyseal dysplasia with White's hypotrichosis (183849, Â): congenital hypotrichosis, rhizomelic short stature, limited abduction of the hips, enlarged metaphyses, delayed ossification of the epiphyses, areas of decay in the metaphyses, vertebral bodies in the thoracic and lumbar regions pear-shaped spine • Strudwick spondyloepimetaphyseal dysplasia (#184250, 12q13.11–q13.2, type II collagen a1 chain gene COL2A1 [120140], Â, the eponym “Strudwick” comes from the surname of one of the patients): severe dwarfism, “chicken” chest", scoliosis, cleft hard palate, retinal detachment, facial hemangioma, inguinal hernia, clubfoot, disproportionately short limbs, normal mental development, sclerotic changes in the metaphyses of long bones, the lesion is greater in the ulna than in the radius and in the fibula more than tibia, delayed maturation of the epiphyses • Spondyloepimetaphyseal dysplasia with joint laxity (*271640, r) • Spondyloepimetaphyseal dysplasia with short limbs (271665, r). ICD-10. Q77.8 Other osteochondrodysplasia with growth defects of long bones and spinal column. OMIM: Spondyloepimetaphyseal dysplasia • Kozlovsky (184252) • White (183849) • Strudwick (184250) • with joint laxity (271640) • with short limbs (271665) • X-linked (300106) • with abnormal dentin development (601668) • type Missouri (*602111) • micromelic (601096).
Spondyloepiphyseal dysplasia is a group of hereditary skeletal diseases that differs from spondyloepiphyseal dysplasias in the absence of damage to the metaphyses of long tubular bones • Congenital spondyloepiphyseal dysplasia (#183900, collagen gene COL2A1 [120140], Â). Клинически: врождённая карликовость с коротким туловищем, нормоцефалия, плоское лицо, миопия, отслойка сетчатки, расщелина твёрдого нёба, платиспондилия, короткая шея, подвывих шейных позвонков, гипоплазия зубовидного отростка, кифоз, сколиоз, поясничный лордоз, цервикальная миелопатия, гипотония, умственная отсталость, barrel-shaped chest, sensorineural hearing loss, hypoplasia of the abdominal muscles, abdominal and inguinal hernias, insufficient ossification of the pubic bones, distal epiphyses of the femur and proximal tibia, talus and calcaneus, flattening of the vertebral bodies • Dysplasia spondyloepiphyseal Maroto (184095, Â): platyspondyly, normal nal intelligence, shortening of the extremities, x-shaped legs deformation, abnormal shape of the input opening of the pelvis • Spondyloepyphysular dystrophy with retinal dystrophy (183850, â) • Spondyloepyphysular dysplasia, myopia and neurosenory hearing loss (184000, â), possibly allel with sticker syndrome • Display of spondyloepyphyphyphyphyphypically Shimke (*242900, r) • Spondyloepiphyseal dysplasia, Irapa type (*271650, r), common among the Irapa Indians in Venezuela and Mexico. Clinically: shortening of the spine, platyspondyly, short bones of the metacarpus and metatarsus, pathological proximal femoral epiphyses and distal humerus • Spondyloepiphyseal dysplasia with atlantoaxial instability (600561, Â) • Spondyloepiphyseal dysplasia pseudoachondroplastic (3 types: 177150, Â; 2641 50, r; #177170 [19p13.1, gene for oligomeric cartilage matrix protein COMP, Â]) is one of the most common skeletal dysplasias. Patients appear normal at birth, and growth retardation is rarely recognized until the second year of life or later. Unlike achondroplasia, the head and face are normal. The fingers are short but do not have the trident shape typical of achondroplasia. There are various deformities of the lower extremities, and ligament weakness is noted. Clinically: short-limb dwarfism, recognized in childhood; lumbar lordosis, kyphosis, scoliosis, dislocations in the atlantoaxial joint, brachydactyly, ulnar deviation of the wrists, limited straightening in the elbow and hip joints, ligament weakness, X-shaped deformity of the legs, chronic myelopathy of the cervical spinal cord, platyspondyly, deformation of the vertebral bodies, shortening of the tubular bones, expansion of metaphyses, abnormal epiphyses • Late dominant spondyloepiphyseal dysplasia (*184100, Â): dwarfism with shortening of the trunk, recognized in childhood, wide face, platyspondyly, short neck, subluxation of the cervical vertebrae, hypoplasia of the odontoid process, kyphoscoliosis, lumbar lordosis, barrel-shaped chest, pathology of the femoral heads with degenerative changes • Late spondyloepiphyseal dysplasia with a characteristic face (600093, r): microcephaly, developmental delay, wide root and tip of the nose, short wide filter (philtrum), thick lips, progressive narrowing of the intervertebral distances, smoothed knee epiphyses • Late spondyloepiphyseal dysplasia with progressive arthropathy (*208230, 6q, PPAC gene, r). Synonym: progressive pseudorheumatoid arthropathy. Clinically: arthropathy, progressive morning stiffness, swelling of the finger joints; histologically: normal synovial membrane, age of onset - about 3 years, reduced mobility of the cervical spine, smoothed vertebral bodies, ossification defects, widened proximal and middle phalanges of the fingers. Laboratory: normal ESR, negative rheumatoid tests, bone dysplasia, pathological acetabulum, short stature in adults (140–150 cm) • Late spondyloepiphyseal dysplasia (*313400, À): congenital dwarfism with short limbs, normal skull shape, flat face, short neck, platyspondyly, subluxation of the cervical vertebrae, hypoplasia of the odontoid process, kyphoscoliosis, lumbar lordosis, barrel chest, degenerative arthritis of the hip joints, diagnosis cannot be made before 4–6 years of age • Late recessive spondyloepiphyseal dysplasia (*271600, r) • Late spondyloepiphyseal dysplasia with mental retardation (271620, r). Clinically: mild or moderate mental retardation, tongue-like shape of the lumbar vertebral bodies, platyspondyly, expansion of the iliac bones, deformity of the acetabulum with hip subluxation and varus deformity in the joint, thin femoral necks. ICD-10. Q77.7 Spondyloepiphyseal dysplasia.
Trichodental dysplasia (601453, Â) - hypodontia and abnormal hair growth. ICD-10. • Q84.2 Other congenital hair abnormalities • K00.8 Other disorders of dental development.
Fibrous bone dysplasia is a violation of the structure of the tubular bone in the form of replacement with fibrous tissue, which leads to its symmetrical curvature and thickening; the process may be limited to one bone or involve many bones (multiple fibrous osteodysplasia) “fibrous osteodysplasia” Lichtenstein–Braitz disease “fibrous osteoma” osteofibroma “local fibrous osteitis. ICD-10. • D48 Neoplasm of undetermined or unknown nature in other and unspecified localizations • D48.0 Bones and articular cartilage.
Frontofacial dysplasia (*229400, frontofacial dysostosis, r) - brachycephaly, cerebral hernia, hypoplasia of the frontal bone, blepharophimosis, ptosis, hare's eye, coloboma of the eyelid and iris, hypertelorism, cataracts, microphthalmos, microcornea, hypoplasia of nasal structures, cleft lip/ palate. ICD-10. Q87.0 Syndromes of congenital anomalies affecting primarily the appearance of the face.
Cranioclavical dysplasia (#119600, 6p21, defect in the CBFA1 transcription factor gene [600211, AML3], Â; 216330, r, severe form). Clinically: moderate growth retardation, brachycephaly, hypoplasia of the middle third of the face, delayed eruption of primary and permanent teeth, supernumerary teeth, spina bifida occulta, widened sacroiliac joints, hypoplasia or aplasia of the clavicles, abnormal position of the shoulder blades, narrow chest, shortened ribs, hypoplasia pubic bones, widening of the symphysis, hypoplasia of the hip joint with dislocation of the hip, brachydactyly, acroosteolysis, joint laxity, syringomyelia, permanently open sutures of the skull with protrusion of the fontanelles, shortening of the middle phalanx of the fifth finger, thin diaphyses of the phalanges and metacarpal bones of the fingers, cone-shaped epiphyses, moderate delay bone age in childhood • Younis-Varon syndrome (*216340, r): large skull with dehiscence, micrognathia, poorly defined lips, absence of clavicles, thumb, distal phalanges of the fingers, hypoplasia of the proximal phalanx of the big toes, pelvic dysplasia, bilateral hip subluxation. ICD-10. Q87.5 Other congenital anomaly syndromes with other skeletal changes.
Epithelial dysplasia of the mucous membranes (*158310, Â). Clinically: damage to the red border of the lips, photophobia, follicular keratosis, nystagmus, keratoconjunctivitis, cataracts, moderate baldness, chronic nail infections, repeated pneumonia, cystic fibrosis lung disease, cor pulmonale, candidiasis of the skin and mucous membranes, diarrhea in infancy, T disorders - and B cellular immunity. Laboratory: in smears from the vagina, oral cavity, urinary tract - large immature cells containing vacuoles and strip-like inclusions, histology of the mucous membranes - dyskeratosis and lack of keratinization, ultrastructure of epithelial cells - lack of keratohyalin, a decrease in the number of desmosomes. ICD-10: coded according to the clinically most significant syndrome for a given treatment.
Reasons for the development of pathology
Experts in the field of dentistry still cannot identify specific reasons that are guaranteed to lead to the formation of a benign tumor on the jaw bone. Today, experts identify several factors that can provoke the development of pathology:
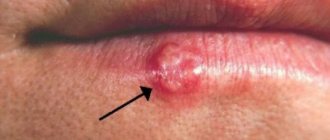
- chronic course of inflammatory processes in the oral cavity - osteomyelitis, actinomycosis, fibrous periodontitis and others,
- severe injury to the jaw bone due to a blow, bruise,
- systematic mechanical damage to hard or soft tissues of the oral cavity - incorrectly installed fillings, dentures, orthodontic apparatus (brackets).
The photo shows osteomyelitis.
Genetic predisposition can also be added to possible predispositions. Negative external influences, such as smoking or radiation therapy, can also contribute to the development of pathology.
Characteristic symptoms
One of the most dangerous characteristics of cementoma is its asymptomatic course. In the first stages of development of the pathology, the patient does not experience any discomfort or pain. Therefore, most often the tumor can only be detected at a dentist’s appointment.
“Dangerous disease! I noticed it already at the last stage, when I went to the dentist to treat a tooth, I just wanted to put a filling and remove one. As a result, they discovered cementoma... And indeed, there was no pain at all, I didn’t feel anything. I’m sure that I’m still very lucky that everything coincided like that...”
Nataly, from correspondence on the forum www.32top.ru
The tumor can be identified by x-ray.
As the size of the tumor increases, the thickness of the cortical plate decreases, which creates unnecessary pressure on the periosteum. At this stage of pathology development, the patient may experience the following symptoms:
- discomfort and moderate pain when eating or talking,
- unpleasant sensations at the moment of mechanical impact on the causative area,
- slight deformation of the enamel structure, change in its color.
When the tumor reaches a large size, it can lead to rupture of the mucous membrane and soft tissues in the oral cavity. In the most advanced cases, there is a risk of deformation of facial features and even fracture of the jaw bone.
Types of dental cementoma - classification
Experts identify several forms of cementoma. Each variety is characterized by its own characteristics and symptoms. Let's look at each type in a little more detail.
Benign cementoblastoma
We are talking about a true cementoma, which is understood as a neoplasm of coarse fibrous tissue with varying degrees of mineralization. The tumor is covered with a kind of film - this capsule separates it from healthy cells. This form is characterized by a rather slow course, but the tumor itself can grow to unlimited sizes. The initial stages of this process are not accompanied by any symptoms, but over time there is pronounced resorption of the cortical plate. As a result, the patient experiences discomfort and unpleasant sensations during mechanical action on the causative area, for example, when brushing teeth or eating.
The tumor is covered by a peculiar capsule
An advanced stage can lead to deformation of the jaw bone and the eruption of a tumor, which is fraught with infection and the development of inflammatory processes. Typically, the tumor occurs on the lower jaw in the premolar area.
Cement-forming fibroma
The tumor grows slowly, and the process of its development is usually accompanied by mild symptoms. A lesion with weak mineralization of the neoplasm is formed in the bone tissue, which is due to its fibroblastic structure. In this case, cement-like tissue is formed already at an advanced stage of the pathology, when tumor growth stops. This form of cementoma is especially dangerous when localized in the upper jaw, where its spread is fraught with damage to the maxillary sinus1.
Periapical cement dysplasia
The pathological process occurs in dental cement and bone tissue, which leads to thinning of the walls of the root system of molars. This situation is dangerous due to root fracture. Cement dysplasia usually has many pathological foci, but all of them, as a rule, do not exceed 1 cm in diameter. Tumor formation is also asymptomatic. In this case, there is no deformation of the jaw. Most often, the neoplasm is localized in the anterior parts of the lower dentition.
Periapical cemental dysplasia is often localized in the mandible
Gigantoform cementoma
This form of cementoma is characterized by intensive development, namely the rapid transformation of connective tissue into cementum. The x-ray image clearly shows oval-shaped pathological foci located in close proximity to the root system of the tooth. In this case, the localization can be very different, but usually tumors arise in places that are symmetrical relative to each other. Many experts agree that giganoform cementoma has genetic background, so often a similar diagnosis is given to several members of the same family.
This variety has oval-shaped lesions
general characteristics
Jaw dysplasia is the replacement of the jaw bone with connective tissue containing elements of cement-like calcifications. The fibrous mass consists of cells responsible for the synthesis of collagen, elastin and other components.
The affected jaw bones lose their normal structure, strength, and their composition and morphology change. The pathological process leads to the emergence of factors:
- Changes in bone shape.
- Facial asymmetry.
- Fractures.
- Destruction of surrounding areas.
- The appearance of inflammation, pain and other clinical signs.
In most cases, the disease is asymptomatic.
The first case of pathology was described by Recklinghausen, a pathologist from Germany in 1891.
There is no consensus among doctors regarding the classification, stages and types of this anomaly. The article describes generally accepted formulations and characteristics.
The localization of foci and the prevalence of the disease are determined by its type. Women are more susceptible to this anomaly compared to men. The average incidence rate is 1-6%.
The key cause is non-hereditary gene mutations that disrupt the normal process of bone formation.
How is diagnostics carried out?
Diagnosis of cementoma is possible only in a dental clinic. It is often possible to identify it after the patient contacts him, but for a different purpose, for example, for the treatment of caries or standard prevention. If, during a visual examination, a specialist develops appropriate suspicions or characteristic symptoms occur, the patient will be sent for an x-ray.
Histology helps to conduct a thorough study of the structure and structure of deformed tissues. The sample is placed on a microslide (slide for the object being studied), after which it is carefully examined in a multiply enlarged format, that is, under a microscope. This allows you to accurately determine the specific form of the tumor and make a final diagnosis. Histological examination also makes it possible to differentiate cementoma from other types of tumors, including malignant ones.
Histological examination is also performed for diagnosis
Advantages of treatment in Israel
- International level of specialist training.
- Modern material and technical base of clinics
- Performing surgical interventions using progressive methods.
- Comfortable conditions for treatment.
- Loyal prices.
Carrying out therapy at the initial stages of the disease allows most patients to forget about the disease and quickly restore lost functions. Do not hesitate, contact an Israeli clinic and undergo a full course of treatment and rehabilitation.
- 5
- 4
- 3
- 2
- 1
(0 votes, average: 5 out of 5)
What treatment is required
The choice of direction of therapy directly depends on the form of the pathology. Thus, benign cementoblastoma and cementing fibroma are treated by surgical removal, including partial excision of the affected area of bone tissue. In this case, the causative tooth has to be removed.
In the case of periapical cement dysplasia, the growth of the tumor does not occur as intensively. The surgical method is practically not used, since such an intervention can create a threat of tissue necrosis. If there are no symptoms, therapy is based on regular observation by a specialist, enhanced oral hygiene, and careful monitoring of the periapical lesion area.
In the presence of pronounced symptoms and the development of the inflammatory process, rejection of the sclerotic masses occurs. This process may take an impressive period of time, but upon completion, complete recovery will be achieved. To speed it up, a specialist can create a small depression in the area of the affected bone.
Conservative methods of therapy are also used to treat gigantoform cementoma. The doctor will definitely prescribe anti-inflammatory drugs and put the patient on regular monitoring.
Modern methods of diagnosing the disease
Diagnosis of this disease, especially its monoosseous form, is difficult. Examination of the patient, consultation with an oncologist, therapist and TB specialist are necessary both to make a diagnosis and to determine the need for treatment or the possibility of replacing it with observation. In Israeli clinics, the stage of diagnosis and selection of the most effective methods of therapy takes about three days.
- Day 1
- Day 2
- Day 3
At the initial consultation with an orthopedist, which the patient must attend after admission to the medical center, the doctor performs a superficial examination, studies the results of previous studies, talks with the patient, collecting a detailed family history, clarifying the nature and duration of the symptoms.
At the final stage of the consultation, the orthopedist-traumatologist draws up a list of required examinations. The next day, the patient undergoes the types of diagnostic examinations indicated in the list of appointments:
- imaging studies (radiography, CT and MRI of the affected bone) - are intended to identify pathology at the earliest stages of development and establish its characteristics;
- bone scintigraphy;
- densitometry - an x-ray research technique that allows you to determine the mineral density of bone tissue, carried out to exclude the presence of multiple pathological foci;
— a biopsy with further morphological analysis is carried out in order to establish the form of the pathology, as well as to determine the exact cause of the disorders, in the absence of indicators sufficient to make a diagnosis after imaging studies.
The results are submitted for consideration to a medical council, which includes an orthopedist and highly specialized specialists. After analyzing the indicators, doctors collectively establish a diagnosis and build a treatment regimen.
Possible complications
Complications most often arise when cementoma is localized in the anterior part of the upper jaw. As the tumor grows, there is a serious risk of its spreading to the nasal sinuses, which can subsequently lead to the development of nasopharyngeal pathologies.
Another possible complication of cementoma may be a breakthrough of the oral mucosa with the tumor coming to the surface. Perforation leads to the appearance of a hole in the bone tissue and mucous membrane. As a result, the risk of infection of the affected tissues, the development of inflammation and the spread of pathological processes to adjacent areas of the jaw increases.
Preventive measures
To minimize the risk of developing cementoma, it is important to ensure proper and complete oral hygiene, and also, if possible, eliminate the risks of accidental injury to the teeth and jawbone. Experts provide a number of specific recommendations in this regard:
- brush your teeth twice a day, rinse your mouth after every meal, use floss and preventative rinses, visit a dental hygienist twice a year to remove plaque and dental deposits,
- promptly seek dental care for injuries to the teeth and mucous membranes,
- do not postpone a visit to the doctor if there is a traumatic factor in the oral cavity, for example, an incorrectly installed filling, crown or prosthesis.
It is worth having professional teeth cleaning twice a year.
Late detection of cementoma can lead to disruption of the integrity of bone tissue, spread of the tumor to healthy parts of the jaw, and the appearance of perforation in the jaw bone and mucous membrane. To avoid all these complications, it is necessary to carefully monitor the condition of your teeth and oral cavity, as well as regularly visit the dentist’s office for preventive maintenance - at least twice a year.
- Trigolos N.N., Makedonova Yu.A., Firsova I.V., Starikova I.V., Piterskaya N.V., Marymova E.B., Poroyskaya A.V. Cement-bone dysplasia of the jaws, 2015.